Alkaptonuria
Dr. Ajay Singh
Expert Insight:
Review by Lina Osama Zaki Quteineh, Consultant Medical Genetics (DHA ID: 9294403)
Unmasking Alkaptonuria: The Genetic Blueprint of Ochronosis and "Black Urine Disease"
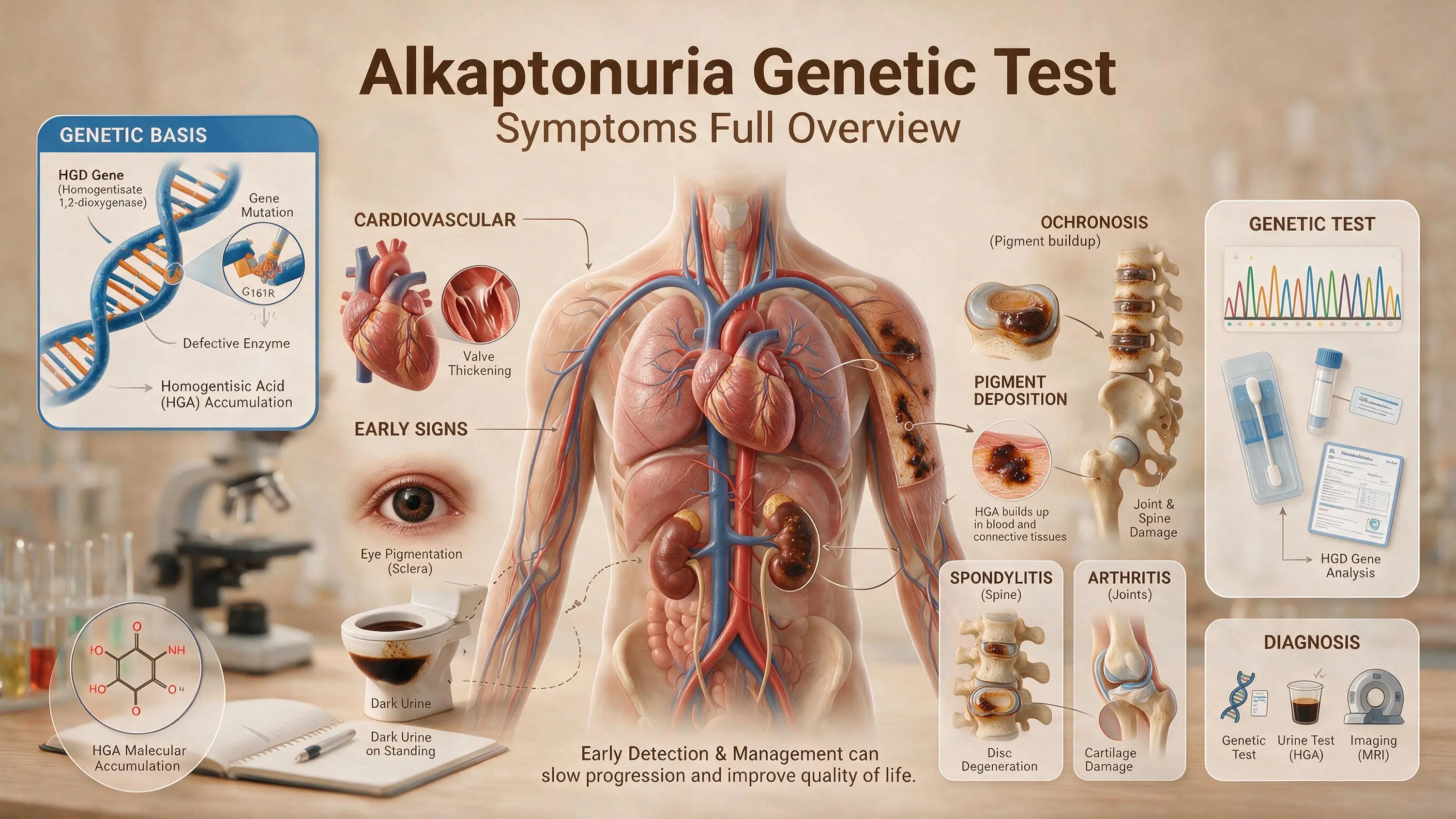
Alkaptonuria (AKU) represents a seminal chapter in the history of medical genetics. First described by Archibald Garrod in the early 20th century as an "inborn error of metabolism," it is the prototypical autosomal recessive disorder . In the UAE, where consanguinity rates are elevated, understanding the genetic blueprint of this condition is particularly relevant for carrier screening and early clinical management.
This condition manifests with a classic triad: dark urine that turns black upon exposure to air, ochronotic pigmentation of connective tissues, and a debilitating arthropathy. However, the clinical trajectory spans decades, often transitioning from a silent biochemical anomaly in childhood to a severe, multi-system disorder in adulthood. Definitive diagnosis relies on full sequencing of the HGD gene, the gold-standard test that identifies the pathogenic variants responsible for this rare condition.
🧬 Physician Insight
"Alkaptonuria is often suspected in childhood when parents notice dark stains in diapers, but the systemic complications—especially ochronotic arthropathy—typically manifest after the fourth decade. Full HGD gene sequencing provides the definitive molecular diagnosis, enabling accurate genetic counseling and timely surveillance for complications such as aortic stenosis and renal stones."
— Lina Osama Zaki Quteineh, Consultant Medical Genetics (DHA Registration ID: 9294403)
The Molecular Pathophysiology of HGD Deficiency
At the center of Alkaptonuria is a deficiency in the enzyme homogentisate 1,2-dioxygenase (HGD), which is primarily expressed in the liver and kidneys . Under normal physiological conditions, this enzyme plays a critical role in the catabolism of the aromatic amino acids phenylalanine and tyrosine.
When structural variants render the HGD gene (mapped to chromosome 3q13.33) non-functional, the metabolic pathway is blocked. This causes a massive accumulation of homogentisic acid (HGA) . While the kidneys filter and excrete substantial volumes of HGA into the urine, residual circulating HGA slowly oxidizes into benzoquinone acetate (BQA). BQA polymerizes into a dense, melanin-like black pigment that binds irreversibly to connective tissues throughout the human body . This systemic tissue staining process is known as ochronosis.
Clinical Evolution: From Infancy to Adulthood
The clinical trajectory of Alkaptonuria spans decades, often transitioning from a silent biochemical anomaly in childhood to a debilitating multi-system disorder in mature adults.
1. Pediatric Manifestations (Homogentisic Aciduria)
During infancy and childhood, AKU is largely asymptomatic, with one definitive exception: the passage of dark urine. When HGA-rich urine is voided, it initially appears completely normal. However, upon exposure to atmospheric oxygen or alkaline agents, the HGA oxidizes, slowly turning the urine dark brown or jet-black. This is frequently first noticed as distinct black stains in infant diapers .
2. Connective Tissue Ochronosis
By the third and fourth decades of life, the chronic accumulation of the BQA polymer manifests visually within specialized anatomical sites :
- The Sclera: Slate-grey or brown pigmentation develops mid-way between the cornea and the inner/outer canthi, typically anchoring at the insertion points of the recti muscles.
- The Auricular Cartilage: The pinna of the outer ear thickens, loses its natural flexibility, and takes on a distinct bluish-black hue.
- Dermal Pigmentation: Sun-exposed regions and areas dense with sweat glands (such as the axillae and forehead) may exhibit a dark, speckled discoloration, occasionally accompanied by chromhidrosis (pigmented sweat that stains garments).
3. Ochronotic Osteoarthropathy
The structural impact on the musculoskeletal system represents the most debilitating phase of the disease. The deposition of HGA polymers into hyaline articular cartilage alters its physical matrix, rendering the cartilage brittle, inflexible, and highly prone to micro-fracturing under normal mechanical stress .
Unlike typical age-related osteoarthritis, ochronotic arthropathy presents primarily as severe, early-onset lumbar pain and spinal stiffness, closely mimicking ankylosing spondylitis . Over time, the intervertebral discs undergo severe calcification and flattening, leading to spinal fusion. Major weight-bearing joints—specifically the hips, knees, and shoulders—experience rapid, severe breakdown that frequently requires total joint replacement surgeries at a young age .
Systemic Extra-Articular Complications
Because collagenous tissues are ubiquitous, Alkaptonuria is inherently a multi-system condition:
Anatomical System Clinical Presentation & Pathology
| Cardiovascular | Pigment deposition within the aortic and mitral valves induces chronic inflammation, leading to severe calcification and aortic valve stenosis. Coronary artery calcification is also heavily accelerated .
| Renal & Urological | The continuous filtration of massive concentrations of HGA precipitates into ochronotic kidney stones and black prostatic calculi, often provoking recurrent urinary tract infections or acute obstructions .
| Endocrine | Clinical registries show a significantly elevated prevalence of metabolic cross-over complications, including hypothyroidism, which requires close endocrine monitoring.
The HGD Gene and Alkaptonuria in the UAE Population
Alkaptonuria is an autosomal recessive disorder caused by mutations in the HGD gene. Over 100 pathogenic variants have been identified, with mutations most frequently affecting exons 6, 8, 10, and 13 .
A landmark UAE-based study screened 2,857 school children and identified a single nucleotide deletion c.342delA in exon 3 (p.Arg58fs) in an affected Emirati family. The parents were first cousins, and the study estimated the allele frequency of alkaptonuria in the UAE at 0.0107 (95% CI: 0.000392–0.03473). The investigators noted that this mutation is ancient, having occurred thousands of years ago, and has spread across a large geographical area .
Given the elevated consanguinity rates in the region, the incidence of Alkaptonuria may be higher than previously estimated. This underscores the importance of pre-symptomatic genetic evaluation, carrier screening, and comprehensive family counseling.
The Critical Role of Genetic and Molecular Screening
Definitive confirmation of Alkaptonuria relies on precise diagnostic validation. While quantitative urinary organic acid analysis via gas chromatography-mass spectrometry (GC/MS) confirms the presence of elevated homogentisic acid, advanced molecular genetic testing provides the definitive biological roadmap.
Full sequencing of the HGD gene identifies the exact homozygous or compound heterozygous pathogenic variants responsible for the enzyme deficiency . This analysis covers all coding exons and flanking intron-exon boundaries, ensuring detection of both common and rare mutations that targeted panels may miss.
In the UAE—where consanguinity rates can run high—pre-symptomatic genetic evaluation, carrier screening, and comprehensive family counseling offer prospective parents vital insights into reproductive risks and open doors for early clinical management strategies.
Understanding your genetic profile allows for targeted surveillance, specialized low-protein dietary management (to restrict the intake of phenylalanine and tyrosine precursors), and timely therapeutic interventions before systemic ochronosis can progress. Nitisinone, a medication that reduces HGA production, has shown promise in treating this condition, making early diagnosis increasingly important .
Expert Oversight & Clinical Governance
Clinical Interpretation: All genetic results are reviewed and signed off by a Consultant Medical Geneticist licensed by the Dubai Health Authority (DHA), ensuring variant classification adheres to ACMG/AMP guidelines.
Corporate Oversight: DNA Labs UAE operates under a DHA-licensed facility (License No. 1143) at Dubai Healthcare City, complying with all federal and local regulations for genetic testing and data privacy.
The Path to a Definitive Diagnosis
Alkaptonuria is often overlooked until ochronotic complications emerge. With the prevalence of the R58fs founder mutation in the UAE population, genetic screening for this condition is particularly pertinent. HGD full gene analysis offers the definitive molecular diagnosis, enabling accurate risk assessment, timely therapeutic intervention, and informed reproductive decision-making for UAE families.
Whether you are a healthcare provider evaluating a patient with early-onset arthropathy or a couple considering preconception carrier screening, understanding the genetic basis of alkaptonuria is essential for preventing and managing this "black urine disease."
References
1. Alkaptonuria (sequence analysis of HGD gene). GTR Test ID: GTR000608911.1. National Center for Biotechnology Information (NCBI).
2. Abdulrazzaq YM, Ibrahim A, Al Khayat AI, Nagelkerke NJ, Ali BR. R58fs mutation in the HGD gene in a family with alkaptonuria in the UAE. Clin Genet. 2008.
3. Ochronosis. StatPearls. National Institutes of Health (NIH). 2023.
4. Ochronosis with Fracture of Intracapsular Neck of Femur and Scoliosis: A Case Report. J Orthop Case Rep. 2023.
5. HGD gene. MedlinePlus Genetics. National Institutes of Health (NIH). 2025.
6. Ochronosis. ScienceDirect. 2012.
⚕️ Medical Disclaimer
This article is for informational purposes only and does not constitute medical advice. Always consult with a qualified healthcare professional for diagnosis and treatment.
Found this helpful? Book a test or consult our team.